Research
We develop theoretical and computational approaches targeting the electronic structure and interactions in extended systems, such as photosynthetic and fluorescent proteins, molecular solids, polymers, and bulk liquids. With these tools, we investigate fundamental aspects of non-covalent interactions and the effect of the environment on electronic structure. This page highlights our recent work in these exciting areas.
Energy and electron transfer in photosynthesis
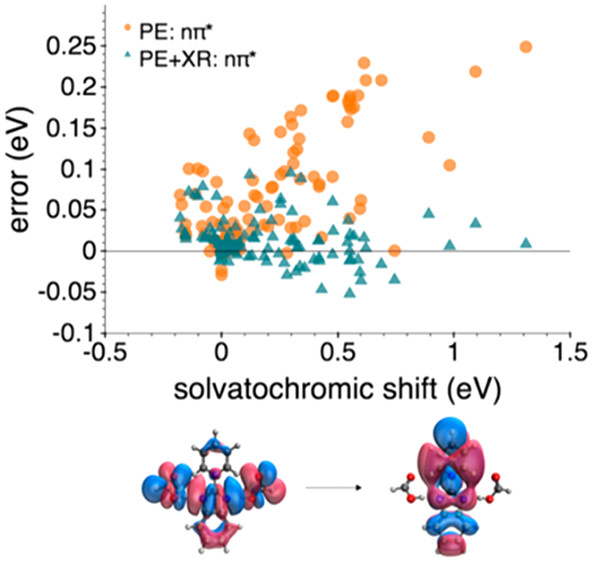
Efficiency and rates of energy transfer in photosystems are controlled by the protein environment. Geometries and relative orientations of chromophores, chlorophylls and carotenoids, are determined by the shapes of protein cavities, whereas electronic excited states of the chromophores is affected by non-uniform electric fields due to the protein. Can we understand and predict the protein effects on the optical spectroscopy and energy transfer in photosynthetic proteins? See below…
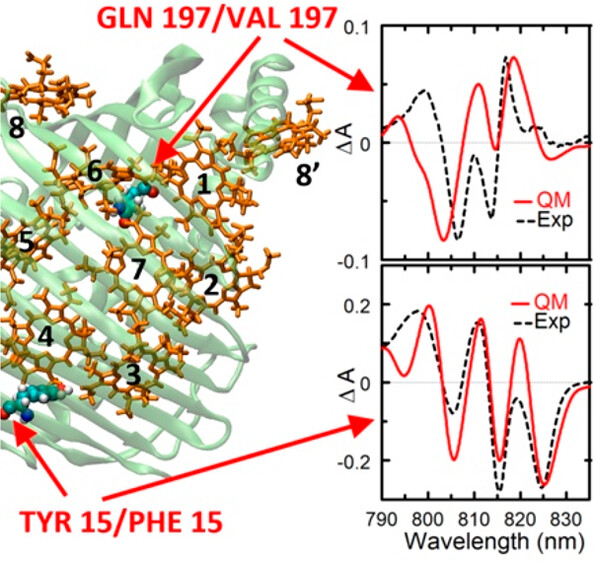
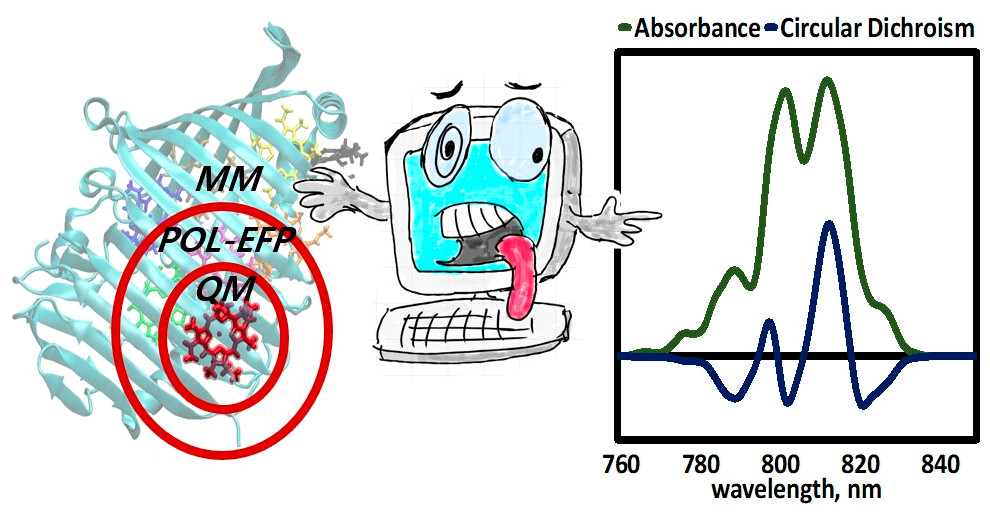
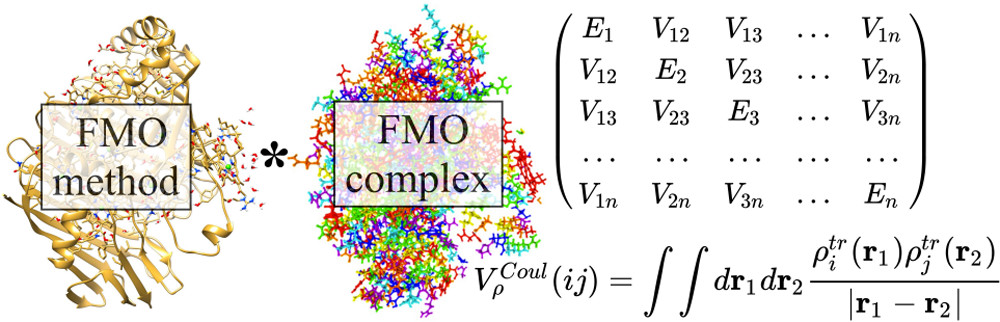
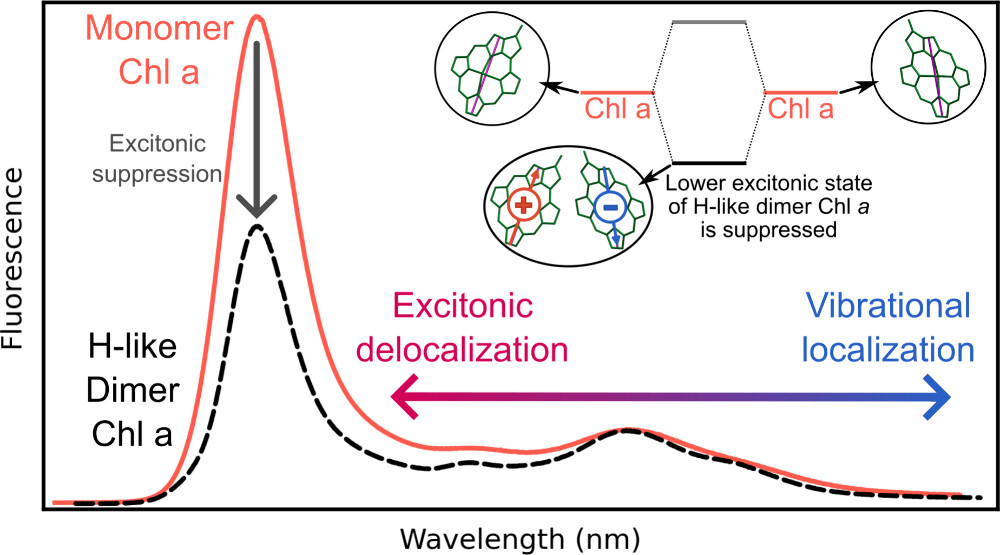
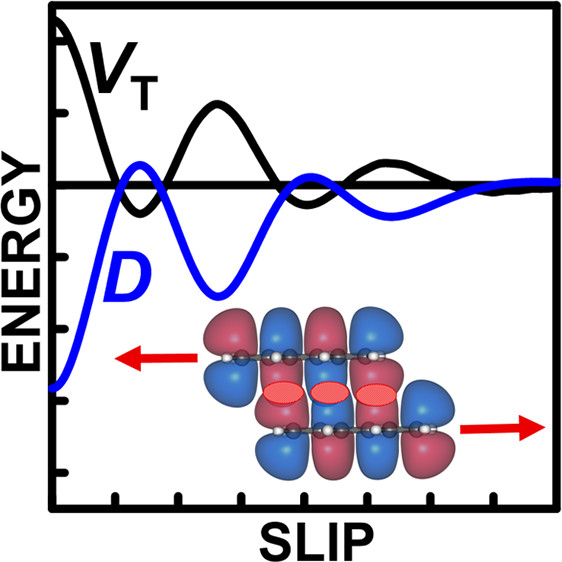
Excitonic and vibronic interactions
Theoretical description of energy transfer in a multi-chromophore systme requires two components, namely:
accurate description of the electronic structure of photoactive pigments
ability to model electronic and vibronic couplings that enable efficient energy transfer between the pigments
The excited-state QM/EFP and FMO methods address the former - see Energy and electron transfer in photosynthesis and Method and software development. To target the second aspect, we develop vibronic models of the Fulton-Gouterman type. Our vibronic model can efficiently treat multiple vibrational modes in multi-chromophore systems.
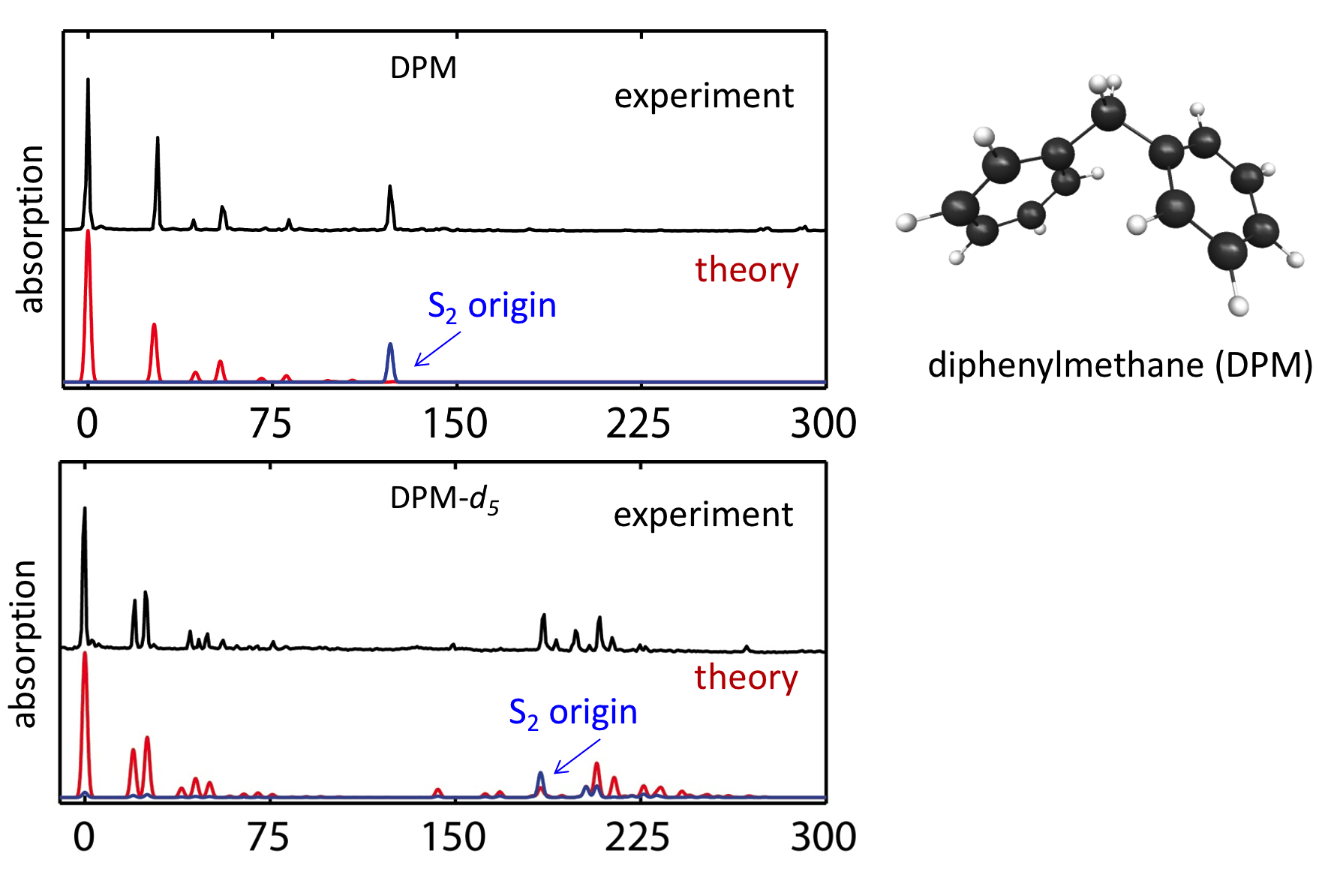
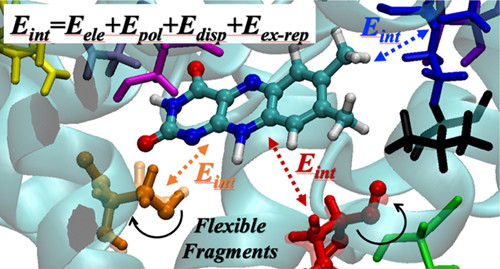
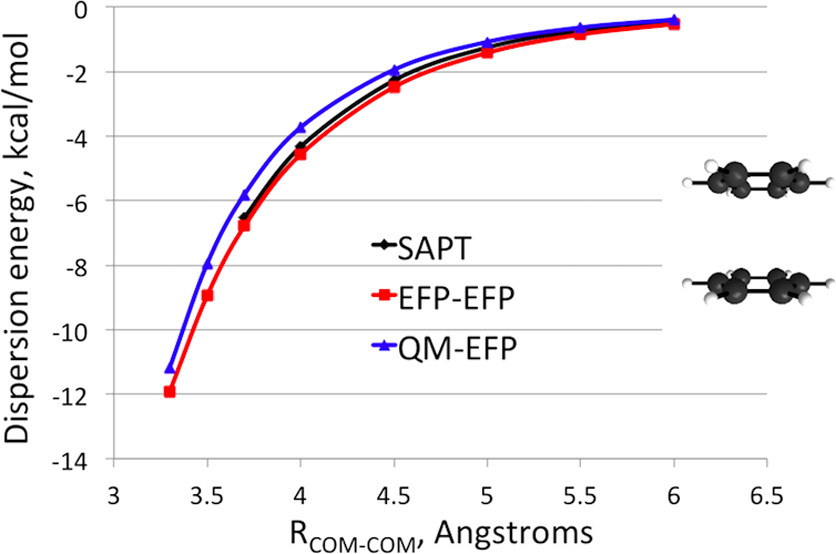
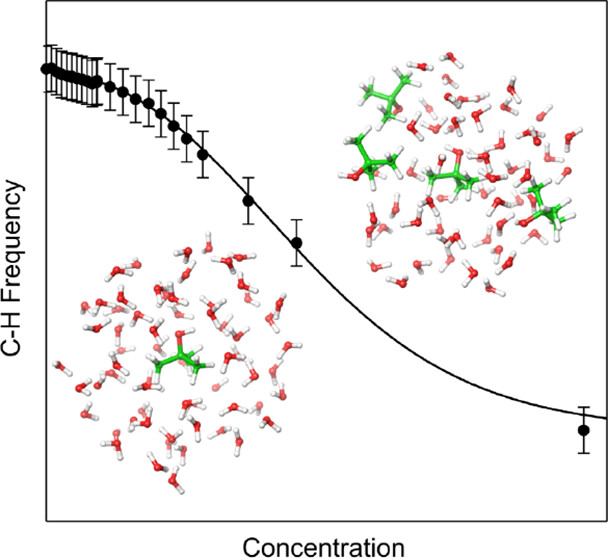
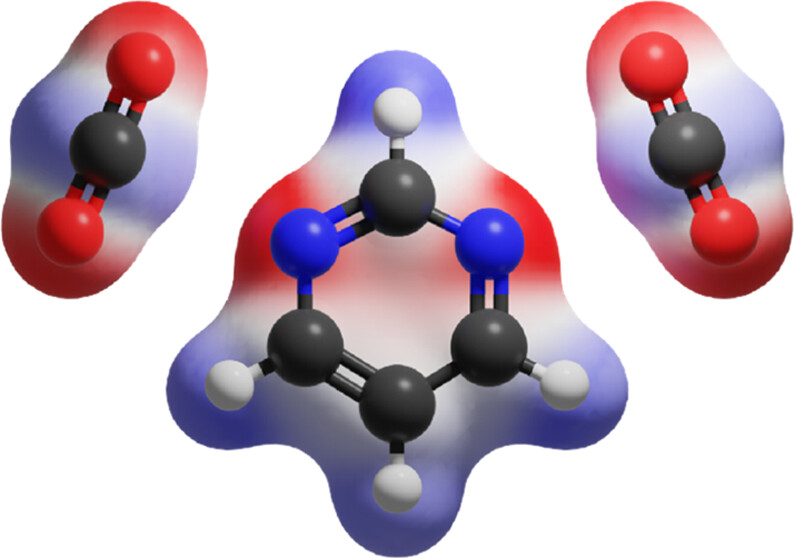
Ubiquitous non-covalent interactions
Non-covalent interactions govern the structure and functions of biological macromolecules such as DNA and proteins, the properties of condensed phase species like liquids and colloids, and adsorption processes of molecules on surfaces and interfaces. We investigate the nature of non-covalent interactions in molecular clusters, heterogeneous liquids, molecular interfaces, and biological systems. In many systems we consider the non-covalent interactions of different types (e.g., electrostatic and dispersive forces) compete with each other. While these situations are challenging for theory and require a balanced description of different non-covalent forces, they provide a rich variety of structural and bonding patterns that exhibit themselves in complicated spectroscopic observables.
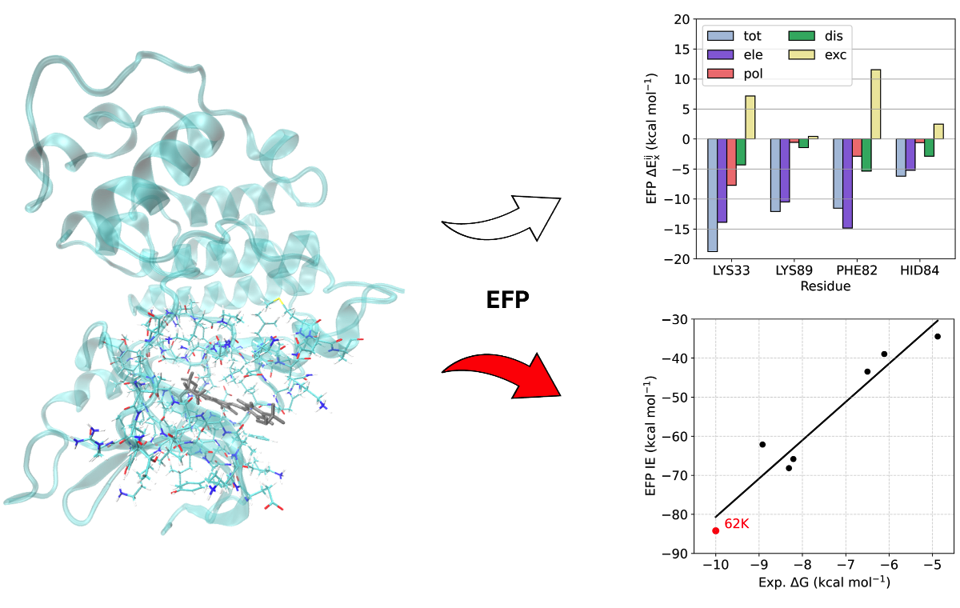
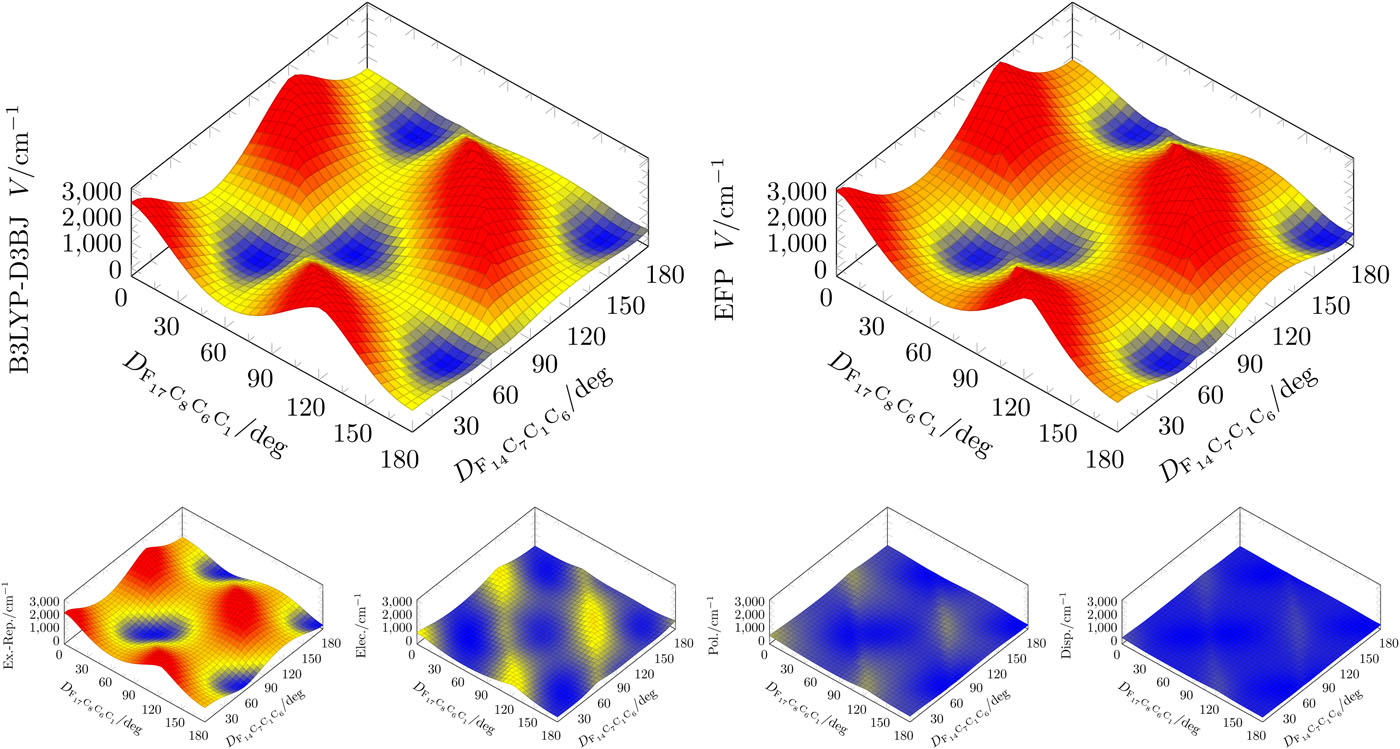
Method and software development
Calculations in the condensed phase still remain a major challenge to the theoretical community. The increased number of nuclear and electronic degrees of freedom makes accurate ab initio calculations of a condensed phase system unfeasible long before the system can approach the bulk. One general approach is then to separate a system into two parts, such that one (active) part is treated by quantum mechanical (QM) techniques, and the other, usually larger, part is calculated by using classical (molecular) mechanics (MM), which is referred to as QM/MM. However, the accuracy of QM/MM models might suffer both from inaccurate force field parameters and intrinsic limitations of the MM force field functional form.
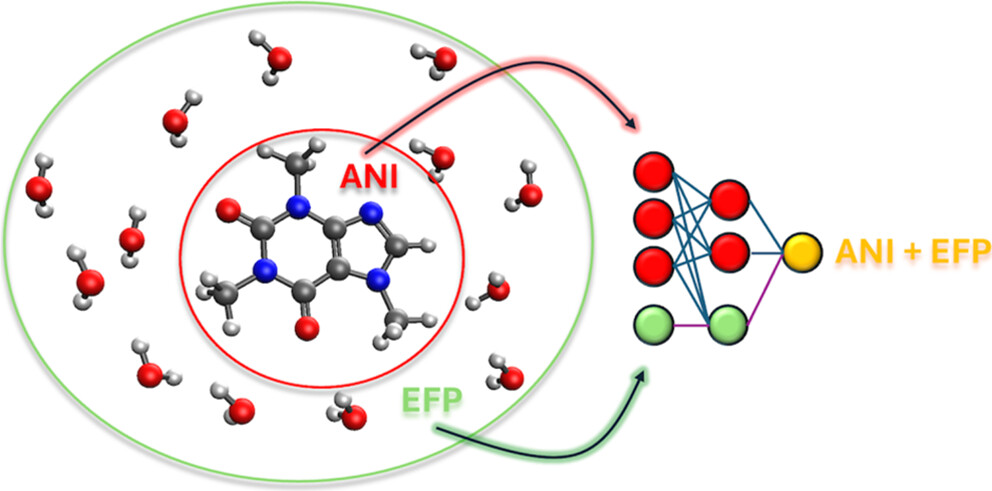
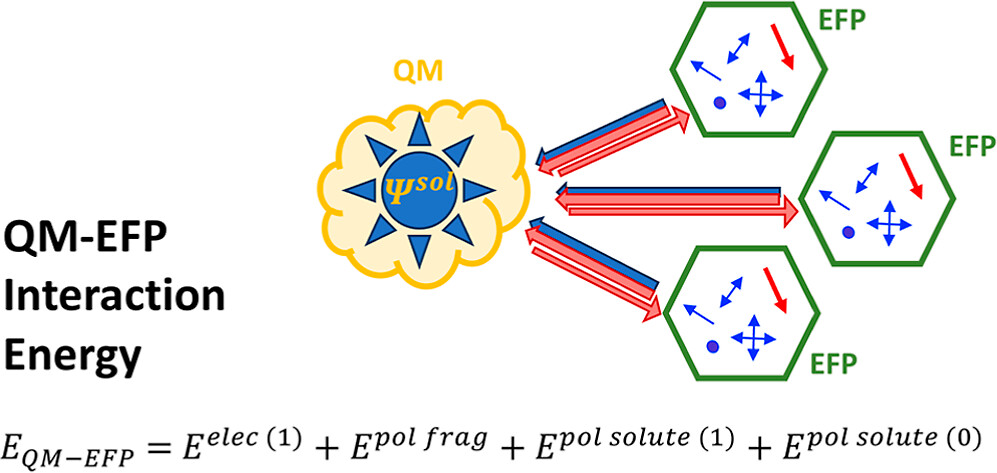
In order to overcome this problem, we utilize the Effective Fragment Potential (EFP) method, which is the ab initio-based polarizable force field.
Our EFP and QM/EFP methods are available in the GAMESS, Q-Chem and Psi4 electronic structure packages, and in our own LibEFP software library.
Note
Check out our EFP website that provides up-to-date information on the EFP development, documentation and tutorials!